|
Case Report
A two-year-old male child with Doose syndrome: An unusual entity
1 Department of Paediatrics, All India Institute of Medical Sciences, Rishikesh, Uttarakhand, India
2 Department of Paediatrics, All India Institute of Medical Sciences, Gorakhpur, Uttar Pradesh, India
Address correspondence to:
Swathi Chacham
Department of Paediatrics, All India Institute of Medical Sciences, Rishikesh, Uttarakhand,
India
Message to Corresponding Author
Article ID: 100077Z06AS2020
Access full text article on other devices

Access PDF of article on other devices

How to cite this article
Singh A, Prabha R, Ahmad N, Chacham S, Mohan K, Gaurav K, Bhat N, Verma PK, Kumar M, Panda PK, Sharawat IK, Prakash B. A two-year-old male child with Doose syndrome: An unusual entity. Case Rep Int 2020;9:100077Z06AS2020.ABSTRACT
Introduction: Myoclonic-astatic epilepsy of early childhood (MAE)/Doose syndrome (DS) is an infrequent entity causing myoclonic-astatic seizures.
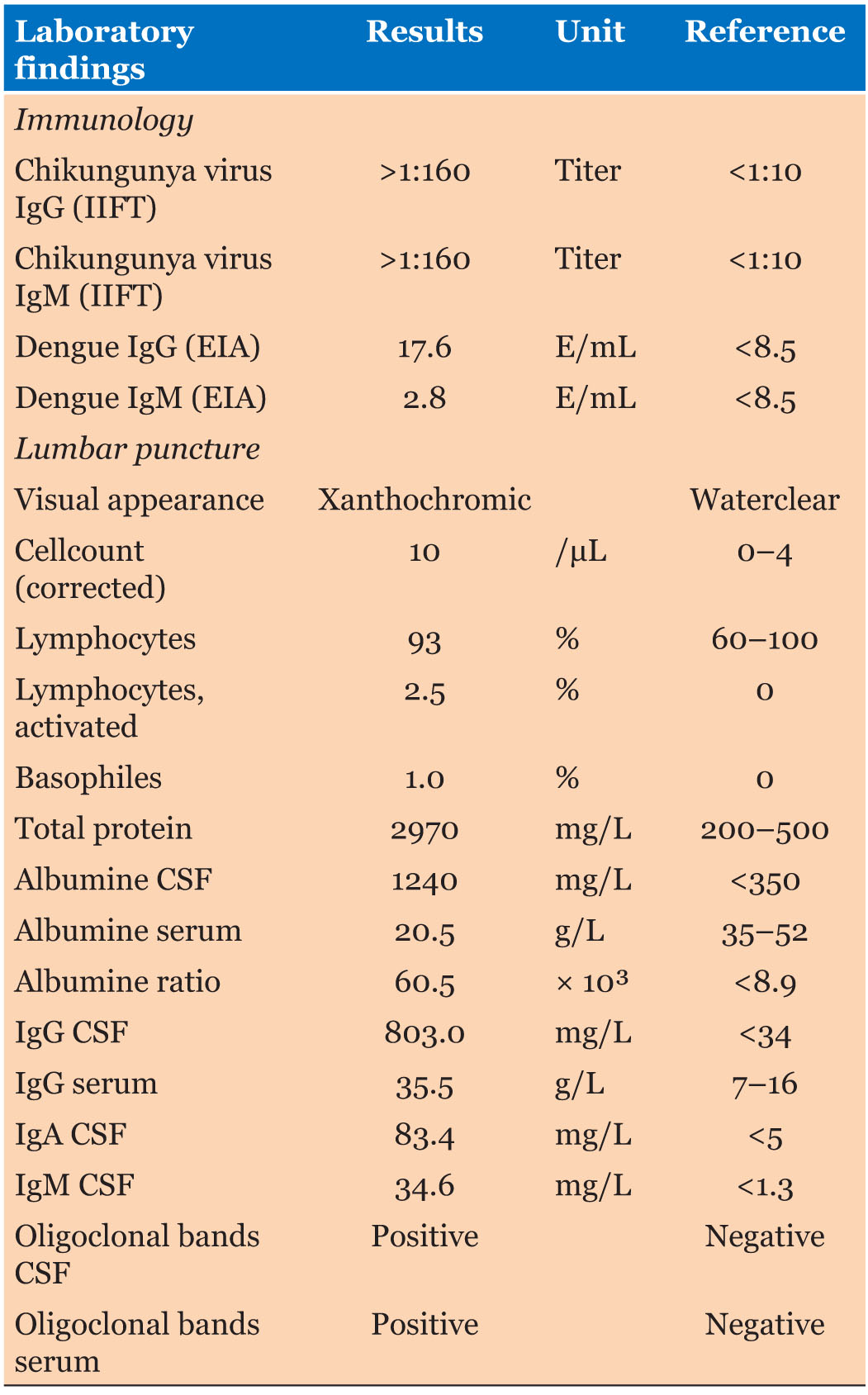
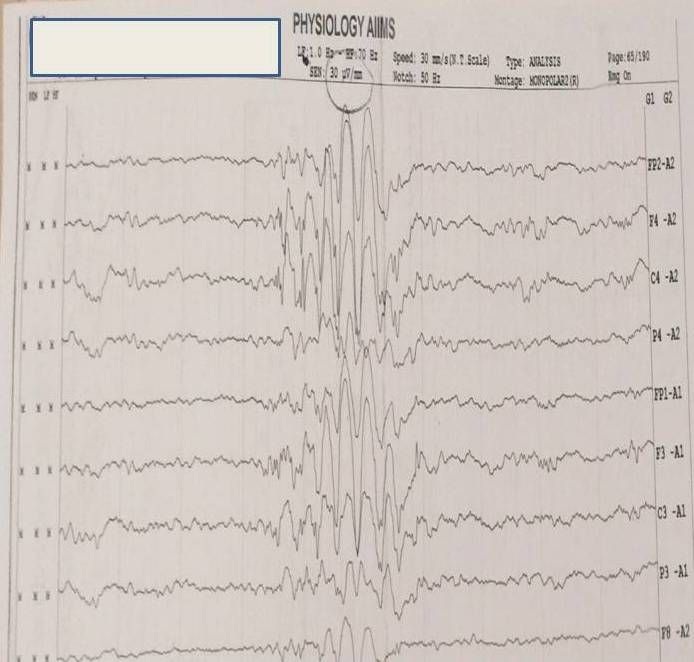
Case Report: A 2-year-old male child presenting with history of seizures from one year, inability to speak and choreiform movements. He had isolated speech delay with other quadrants being normal (speaks monosyllables at two years). Parents noticed brief jerky movements of both upper and lower limbs with increasing intensity over 2–3 months which became a daily occurrence. Also, the child had episodes of abrupt loss of contact with surroundings and prolonged staring (3–5 minutes) and few eye blinks. Subsequently, by 1.5 years, he started having episodes of sudden forward bending of head followed by frequent falls. Jerks either preceded or followed falls. Decreased attention and hyperactivity had started by one year of the illness. Electroencephalography depicted polysharp waves with maximum voltage of 180 μV and 3–4 Hz@ an interval of 20–30 seconds bilaterally. He responded to oral sodium valproate, levetiracetam, and ketogenic diet.
Conclusion: We report a 2-year-old male child with Doose syndrome, a distinct yet rare entity.
Keywords: Astatic seizures, Atonic seizures, Electroencephalographic correlate, Myoclonic-astatic epilepsy, Myoclonic-atonic seizures, Myoclonic seizures
INTRODUCTION
Doose syndrome also known as myoclonic-astatic epilepsy of early childhood (MAE), a rarely diagnosed entity, a kind of cryptogenic epilepsy with myoclonic-astatic seizures as per the International League against Epilepsy (ILAE). Although accounting for 1–2% of all epilepsies, it is often underdiagnosed. Recent ILAE classification [1],[2],[3],[4],[5],[6], recognizes four distinct epileptic syndromes including MAE, benign and severe myoclonic epilepsy in infants [7],[8], and cryptogenic variant of Lennox–Gastaut syndrome (LGS) [9]. Forty years after description of this entity by Herman Doose in 51 children, the clinical and electrographic features of Doose syndrome (DS) have been well characterized. Its differentiation from other epileptic syndromes such as Dravet syndrome, LGS, progressive myoclonic epilepsy, and Landau–Kleffner syndrome (LKS) is vital. Despite a number of publications regarding the clinical and electroencephalographic (EEG) characteristics among the former three syndromes, MAE has been underreported. Syndromic classification enables physicians to predict outcomes as well as select treatments specific for the syndrome. Considering the fact that half of MAE patients might run an unfavorable clinical course, it is crucial to identify better treatment strategies and expected outcomes of this epileptic syndrome. Genetic factors play an important role with clinical seizures or subclinical EEG abnormalities being reported in family members of Doose syndrome in 40–80% of cases. We report a 2-year-old male child with Doose syndrome and polymorphic epilepsy.
CASE REPORT
A 2-year-old male child was brought to OPD with history of seizures along with delayed speech and choreiform movements since one year of age. He was born of non-consanguineous marriage with uneventful antenatal, natal, and postnatal period. He had isolated speech delay with other developmental quadrants being normal (speaks monosyllables at two years). Parents noticed brief jerky movements involving both upper and lower limbs whose intensity increased over 2–3 months and the frequency increased so much so that it became a daily occurrence. Also, the child had episodes of sudden loss of contact with surroundings and prolonged staring (3–5 minutes) episodes and few eye blinks. There was no history of behavioral abnormalities. Subsequently, by 1.5 years, he started having variable presentation of sudden forward bending of head followed by frequent falls. Jerks either preceded or followed falls. There were no recognizable precipitating factors such as hot bath, cold bath, sleep or sleep deprivation, fever, bright lights, or noise for any of the seizure types. The child continued to have normal cognitive abilities in the interictal period. However, decreased attention and hyperactivity had set in about one year of the illness. There was no family history of similar complaints.
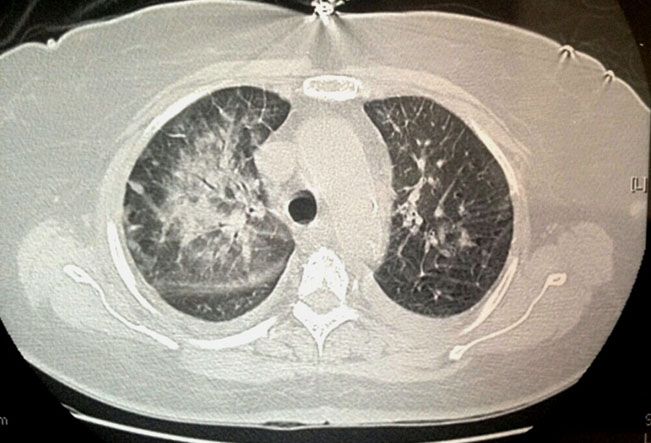
Examination (as shown in Figure 1) revealed normal head size, copper colored hair with dental dysplasia. However, no neurocutaneous markers were seen and no autistic activities observed. Cranial nerve examination, motor, sensory, and cerebellar assessment were unremarkable. Also, other systemic examination was normal. Electroencephalography done under sedation revealed (as shown in Figure 2) polysharp waves with maximum voltage of 180 μV with a frequency of 3–4 Hz@ an interval of 20–30 seconds bilaterally. Interestingly, right left symmetry was normal throughout the EEG and brain imaging was normal.
Treatment
The child was initially started on oral sodium valproate at 20 mg/kg and later on levetiracetam (@20 mg/kg/day) was added. He was also started on. The parents were advised to provide the child with substances rich in fat like easily available cream, cheese, butter, ghee, and egg and avoid carbohydrate rich foods like candies, cookies, and sweets (ketogenic diet). As the parents were receptive and understood the need for dietary modification, they continued it on a regular basis. Although it was difficult to withhold candies and sweets from the child, he was pacified with other eatables in place of them. With these therapeutic measures, the child had shown significant decrease in frequency of seizures in the form of seizure episodes once in six months from about two seizures per week. He was on frequent follow-up with good control of seizures.

DISCUSSION
Doose syndrome is a form of childhood epilepsy with polymorphic seizure episodes including classical myoclonic-astatic or myoclonic-atonic seizure along with other seizure types, such as absence, complex partial, and generalized tonic-clonic seizures. However, it is distinct from related syndromes such as LGS, Dravet syndrome, and LKS with affected individuals having a normal or only a mildly affected cognition and normal development [2].
Myoclonic-atonic seizure is the classical seizure type in DS where the patient suddenly drops to the ground but the falls may be difficult to distinguish from those precipitated by severe myoclonic jerks. These falls are usually time-locked to a generalized spike and wave or polyspike and wave discharges in the EEG [3].
Myoclonic, tonic-clonic, absence, and complex partial seizures can occur concurrently. EEG findings in DS can vary from normal to marked slowing and generalized spike wave discharges. Subclinical EEG abnormalities and abnormal responses to hyperventilation and photic stimulation may be found in family members without overt seizures. Electroencephalography usually demonstrates background frequencies with slow waves without any K complexes or sleep spindles [3].
Various associations have been projected in this syndrome with polygenetic and multifactorial inheritance like neuronal sodium channel 1 (SCN1) and GEFS+ syndrome [4]. In the management, polytherapy is often needed with valproate, levetiracetam, topiramate, and lamotrigine being the commonly used antiepileptic drugs [5]. Ketogenic diet and immunotherapy with adrenocorticotropic hormone (ACTH) and steroids have also been found to be effective in refractory seizures [6]. Newer modalities of treatment include voltage-gated potassium channel antagonists and corpus callosotomy.
Children with epilepsy among 1–5 years age group usually exhibit multiple seizure types thereby raising diagnostic problems. Semiological description and syndrome classification remain bizarre owing to overlap of seizure semiology among various epileptic syndromes of this age. Among patients with Doose syndrome or LGS, polymorphic seizure types including febrile (FS), clonic (CS), myoclonic (MS), myoclonic astatic (MAS), generalized tonic-clonic (GTCS) and absence (AS) seizures can occur. Other epileptic syndromes like severe myoclonic epilepsy of infancy (SMEI, Dravet syndrome) and atypical benign partial epilepsy/pseudo-Lennox syndrome (ABPE/PLS) with continuous spike and waves during slow sleep (CSWS) can have a similar presentation. It was first described by Harper who reported 14 children with true myoclonic epilepsy [10], many reviews have been published ever since. In them, he demonstrated astatic features following violent myoclonic attacks, which were usually resistant to antiepileptic drugs (AEDs). Detailed description of 51 children with so-called centrencephalic myoclonic-astatic petit mal seizures was given by Doose [6], which was later renamed as myoclonic-astatic epilepsy [8],[9]. Benign myoclonic epilepsy in infants (BME) [7],[11]and severe myoclonic epilepsy in infants (SME) [8],[12],[13] were established as distinct epileptic syndromes as proposed by Dravet, which are now accepted by ILAE along with MAE described by Doose [9].
Modified ILAE criteria for MAE include: (1) A child who is developmentally normal prior to the beginning of epilepsy without any organic cerebral abnormalities; (2) Seizures occurring between 7 months and 6 years of age; (3) Generalized spike or polyspike-wave EEG discharges at 2–3 Hz without focal spike discharges; and (4) Exclusion of BME, SME, and LGS. However, currently there is no diagnostic test for MAE, which remains in essence an electro-clinical diagnosis.
Drug therapy
Valproic acid and ethosuximide were recommended in combination for MS/AS and primidone for GTCS [8] by Doose, along with ACTH for minor epileptic status. For severe form of MAE resembling SME, bromide can also be tried [14]. Better responses to ACTH and ketogenic diet are reported in comparison to ESM, as both treatments have proved to be equally effective for atypical absence seizures and myoclonic seizures but not generalized tonic seizures (not usually seen in MAE) [15],[16].
Prognosis
Outcomes can range from normal cognition to severe intellectual disability and from seizure free period to intractable seizures. Predicting the outcome remains difficult in the first year of disease, either clinically or from the EEG findings. Markers of unfavorable prognosis include: cognitive decline, status epilepticus, GTCS in the first two years of life, early initiation of myoclonic status, persisting abnormal theta rhythm, and failure developing alpha rhythm in the background.
About 80–90% of children with Doose syndrome may exhibit normal cognition or only minimal cognitive impairment. As a hallmark of MAE, it usually subsides within 1–3 years of the onset. Almost two-thirds of minor epileptic status disappear. Generalized tonic-clonic seizures tend to continue, especially during sleep.
This case demonstrates a male child with recurrent falls secondary to myoclonic jerks associated with loss of postural tone. Associated EEG are bursts of 2–4 Hz spike and polyspike epileptiform activity, typical of MAE [3]. Modified Atkin’s diet (ketogenic diet) has been particularly helpful, with almost 50% decrease in the occurrence of seizures [4]. Topiramate has also been reported of having good efficacy and is a good drug for refractory atonic seizures that can be administered for a long durations [6],[7]. Paradoxical exacerbation of seizures have been demonstrated with levetiracetam [8]. Protective measures must be incorporated in view of sudden falls which can lead to injuries. Adrenocorticotropic hormone parenterally might be effective in refractory cases. Intractable epilepsy with intellectual impairment seems to be the inevitable outcome in many patients. School absenteeism can also be the cause for intellectual deficit.
CONCLUSION
We report a case of a 2-year-old male child with DS, a unique entity. It is a distinct clinical syndrome with a familial preponderance as well as occasional clustering of cases. The recognition of this entity and differentiation from other related electro-clinical syndromes can sometimes be challenging.
REFERENCE
1.
Aicardi J, Chevrie JJ. Myoclonic epilepsies of childhood. Neuropadiatrie 1971;3(2):177–90. [CrossRef]
[Pubmed]

2.
Kelley SA, Kossoff EH. Doose syndrome (myoclonic-astatic epilepsy): 40 years of progress. Dev Med Child Neurol 2010;52(11):988–93. [CrossRef]
[Pubmed]
3.
Aicardi J. Lennox-Gastaut syndrome. In: Aicardi J, editor. Epilepsy in children. New York: Raven Press; 1992. p. 44–66.
4.
Oguni H, Tanaka T, Hayashi K, Funatsuka M, Sakauchi M, Shirakawa S, Osawa M. Treatment and long-term prognosis of myoclonic-astatic epilepsy of early childhood. Neuropediatrics 2002;33(3):122–32. [CrossRef]
[Pubmed]
5.
Dalla Bernardina B, Capovilla G, Chiamenti C, Trevisan E, Colamaria V. Cryptogenic myoclonic epilepsies of infancy and early childhood: Nosological and prognostic approach. In: Wolf P, Dam M, Janz D, Dreifuss FE, editors. Advances in Epileptology. Volume 16. New York: Raven Press; 1987. p. 175–9.
6.
Doose H, Gerken H, Leonhardt R, Völzke E, Völz C. Centrencephalic myoclonic-astatic petit mal. Clinical and genetic investigations. Neuropadiatrie 1970;2(1):59–78. [CrossRef]
[Pubmed]
7.
Dravet C, Bureau M, Oguni H, Fukuyama Y, Cokar O. Severe myoclonic epilepsy in infancy. (Dravet syndrome). In: Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. 4ed. London: John Libbey Eurotext Publishers; 2005. p. 89–13.
8.
Dravet C, Bureau M, Guerrini M, Giraud N, Roger J. Severe myoclonic epilepsy in infants. In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. 2ed. London: John Libbey; 1992. p. 75–88.
9.
Proposal for revised classification of epilepsies and epileptic syndromes. Commission on classification and terminology of the international league against epilepsy. Epilepsia 1989;30(4):389–99. [CrossRef]
[Pubmed]
10.
Snead OC 3rd, Hosey LC. Treatment of epileptic falling spells with ethosuximide. Brain Dev 1987;9(6):602–4. [CrossRef]
[Pubmed]
11.
Giovanardi Rossi P, Gobbi G, Melideo G, Parmeggiani A, Tullini A, Santucci M. Myoclonic manifestations in the Lennox-Gastaut syndrome and other childhood epilepsies. In: Niedermeyer E, Degen R, editors. The Lennox-Gastaut Syndrome. New York: Alan R. Liss 1988. p. 137–58.
12.
Dulac O, Plouin P, Shewmon A. Myoclonus and epilepsy in childhood: 1996 Royaumont meeting. Epilepsy Res 1998;30(2):91–106. [CrossRef]
[Pubmed]
13.
Guerrini R, Dravet C, Gobbi G, Recci S, Dulac O. Idiopathic generalized epilepsies with myoclonus in infancy and childhood. In: Malafosse A, Genton P, Hersch E, Marescaux C, Broglin D, Bernasconi R, editors. Idiopathic Generalized Epilepsies: Clinical, Experimental and Genetic Aspects. London: John Libbey; 1994. p. 267–80.
14.
Oguni H, Hayashi K, Oguni M, et al. Treatment of severe myoclonic epilepsy in infants with bromide and its borderline variant. Epilepsia 1994;35(6):1140–5. [CrossRef]
[Pubmed]
15.
Kaminska A, Ickowicz A, Plouin P, Bru MF, Dellatolas G, Dulac O. Delineation of cryptogenic Lennox–Gastaut syndrome and myoclonic astatic epilepsy using multiple correspondence analysis. Epilepsy Res 1999;36(1):15–29. [CrossRef]
[Pubmed]
16.
Berth SH, Kossoff EH. Adolescent-onset absence epilepsy years after resolution of childhood epilepsy with myoclonic-atonic seizures. Epilepsy Behav Rep 2019;12:100329. [CrossRef]
[Pubmed]
SUPPORTING INFORMATION
Author Contributions
Rashmie Prabha - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Avinish Singh - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Najeeb Ahmad - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Swathi Chacham - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Kriti Mohan - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Kumar Gaurav - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Nowneet Bhat - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Prashant Kumar Verma - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Manish Kumar - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Prateek Kumar Panda - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Indar Kumar Sharawat - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Brahm Prakash - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Guarantor of SubmissionThe corresponding author is the guarantor of submission.
Source of SupportNone
Consent StatementWritten informed consent was obtained from the patient for publication of this article.
Data AvailabilityAll relevant data are within the paper and its Supporting Information files.
Conflict of InterestAuthors declare no conflict of interest.
Copyright© 2020 Avinish Singh et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information.