| Table of Contents | |
|
Case Report
| ||||||
| Unusual presentation of a retinoblastoma: A rare case | ||||||
| Luvo Gaxa1, Bafana Elliot Hlatshwayo2 | ||||||
|
1MBChB, Registrar, Diagnostic Radiology and Imaging, Polokwane-Mankweng Hospital Complex, Polokwane, Limpopo, South Africa.
2Dip Rad Diag, MBChB, MMed Rad D, Senior specialist, Diagnostic Radiology and Imaging, Polokwane-Mankweng Hospital Complex, Polokwane, Limpopo, South Africa. | ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article]
[Similar article in Pumed] [Similar article in Google Scholar] |
| How to cite this article: |
| Gaxa L, Hlatshwayo BE. Unusual presentation of a retinoblastoma: A rare case. Case Rep Int 2015;4:30–33. |
|
Abstract
|
|
Introduction:
Although a retinoblastoma is a well recognized condition it is still associated with a high mortality rate especially in developing countries because partly patients tend to present at very late stages to the health facilities. The mean age of occurrence of a retinoblastoma is 18 months and the age range of a retinoblastoma occurrence is 0–5 years. Diagnosing a retinoblastoma above the age of six years is extremely rare and exceptional.
Case Report: An eight-year-old girl was presented with a six-month history of a fast growing right eye mass and proptosis. The mass was fungating and septic. The patient had normal developmental milestones. At presentation the body weight 21.40 kg.The vital signs were within normal limits (temperature 36.8°C, pulse 83 beats/min, blood pressure 116/77 mmHg). The diagnosis of a retinoblastoma was highly suspected on clinical and radiological imaging but due to patient's age there was also a suspicion of a rhabdomyosarcoma of the orbit. Biopsy was performed and the diagnosis of a retinoblastoma was confirmed and the patient was then started on chemotherapy. The patient later demised while on treatment due to a disease progression. Conclusion: A retinoblastoma shows some typical imaging findings on computed tomography scan and on magnetic resonance imaging scan, and a multi-disciplinary approach helps greatly to secure the diagnosis and that helps improve the patient's management and prognosis. | |
|
Keywords:
Childhood cancer, Children, Infant, Tumor, Retinoblastoma
| |
|
Introduction
| ||||||
|
Retinoblastoma is a rare malignant intra-ocular tumor that arises in the retinal neuro-ectodermal cells of infants and children and the angiogenic potential of the tumor correlates well with its aggressive nature [1] [2]. Retinoblastoma is estimated to represent 3% of all childhood cancers and is a blinding, devastating and a life-threatening disease in the pediatric population [3] [4]. The estimated incidence of the retinoblastoma ranges from 1 in 15000 to 1 in 20000 live births and the mean age at diagnosis is 18 months [5] [6]. This case is presented because it falls within the extremely rare spectrum of retinoblastoma presentation which is known to present commonly up to five years of age in literature. | ||||||
|
Case Report
| ||||||
|
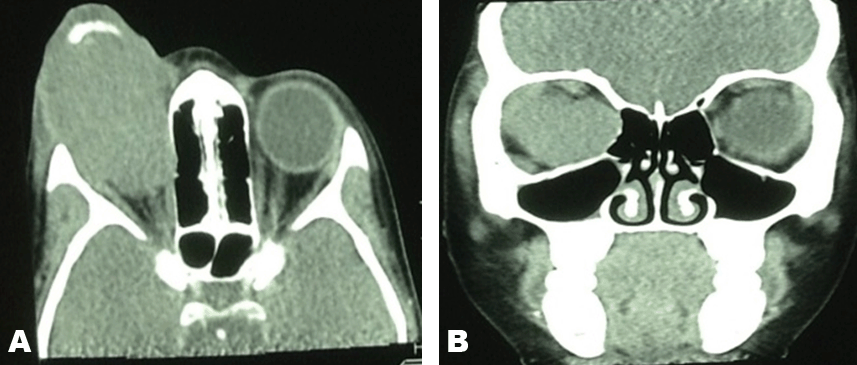
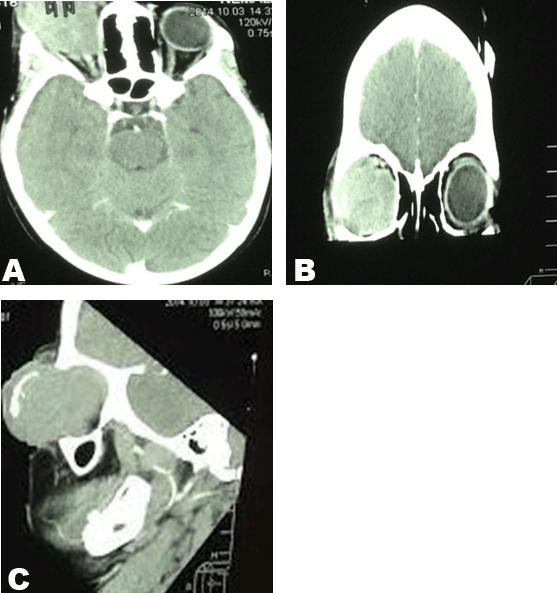
An eight-year-old girl was presented with a six-month history of a fast growing right eye mass and proptosis. The mass was fungating and septic. The parents reported that there was no known past medical history of the patient. The birth history showed that the patient was born through a normal vaginal delivery with an uneventful birth history. The birth weight was 3.1 kg and 48 cm in length. The Apgar score was 10/10. The patient had normal developmental milestones. At presentation the body weight 21.40 kg. The vital signs were within normal limits (temperature 36.8°C, pulse 83 beats/min, blood pressure 116/77 mmHg). The complete blood count results were as follows: Urea and creatinine results were within normal limits. The liver function tests, ESR and the CRP results were also within normal ranges except for the lactate dehydrogenase that was markedly elevated at 1216 U/L while the maximal normal reference range is 295 U/L. Computed tomography scan showed an aggressive right orbital soft tissue mass that had ruptured the right globe with extension of the mass to the surrounding soft tissue and the area of the right optic nerve with no intra-cranial extension. Significantly, the anterior aspect of the mass demonstrated a large calcification. Biopsy was performed and the biopsy results confirmed a retinoblastoma. Microscopic examination: Sections demonstrated conjunctival tissue within which there is an underlying infiltrating malignant small round blue cell tumor. The tumor is arranged as sheets of cells with a rim of basophilic cytoplasm, vesicular nuclei and occasional prominence of nucleoli. There is brisk mitotic activity and apoptosis. The lesion demonstrated numerous vestibule Flexner-Wintersteiner rossettes. The tumor extends into the overlying stratified squamous epithelium and there is associated ulceration and mixed inflammation and the above mentioned features are those of a retinoblastoma. The patient was then started on chemotherapy and the chemotherapy medication that was given was a 1.2 mg of Vincristine intravenously and Stat, 120 mg of etoposite in 200 ml of normal saline over 2 hours for 2 days and carboplatine 240 mg in 200 ml 5% over 2 hours for 2 days. Initially, the patient appeared to have tolerated chemotherapy very well but due to disease progression the child died later while she was in the continuous course of chemotherapy. | ||||||
|
| ||||||
| ||||||
|
Discussion
| ||||||
|
A retinoblastoma is a rare malignant intra-ocular tumor that arises in the retinal neuro-ectodermal cells of infants and children and the angiogenic potential of the tumor correlates well with its aggressive nature [1] [2]. Retinoblastoma is estimated to represent 3% of all childhood cancers and is a blinding, devastating and a life-threatening disease in the pediatric population [3] [4]. Retinoblastoma is the eye carcinoma that develops rapidly and can either be heritable or inheritable [7]. A heritable retinoblastoma typically presents in younger patients commonly younger than the age of one year whereas the inheritable type presents in patients who are older than the age of one year [8] [9]. The incidence of subsequent neoplasms is markedly increased in patients with a heritable form of retinoblastoma and melanoma is the most common of the subsequent neoplasms in long-term retinoblastoma survivors [10]. Familial history of retinoblastoma is noted in 10% of patients and in familial retinoblastoma the germ-line is transmitted from generation to generation and the pathology results from that the mutation has occurred in some ancestor [2] [11]. The estimated incidence of the retinoblastoma ranges from 1 in 15000 to 1 in 20000 live births and the mean age at diagnosis is 18 months [5] [6]. The unilateral cases of retinoblastoma are commonly diagnosed at 24th month of age while the bilateral cases are diagnosed before the age of 12 months [6]. Children are diagnosed with retinoblastoma between the ages of 0–5 years of age and it is known that 80% of retinoblastoma cases are diagnosed prior the age of three years while 95% of retinoblastoma cases are diagnosed before the age of 5 years and reports are scanty regarding a retinoblastoma occurring after the age of six [12]. The latter statement makes the case we presented extremely rare and exceptional because in our patient a diagnosis of a retinoblastoma was made for the very first time at the age of eight years. Retinoblastoma presents with calcifications in 90% of cases which sets this tumor apart from the rest of the eye pathologies which entail but not limited to toxocariasis, Coat's disease, persistent hyperplasic primary vitreous and retrolental fibroplasia [2] . An ophthalmologist performs both fundoscopy and ultrasound to diagnose a retinoblastoma and in almost all cases of retinoblastoma an intra-tumoral calcification is evident on ultrasound which a high confidence level pertaining to the diagnosis [13] . Evaluation of a retinoblastoma for a laterality, tumor size and localization, number and tumor seeding can be confidently achieved with fundoscopy and ultrasound [13]. It is of utmost importance noting that the management of a patient with a retinoblastoma needs a multi-disciplinary co-operation which involves radiologists, pediatric oncologists, radiation oncologists, ophthalmologists and many other related specialities [14]. The danger of not treating a retinoblastoma is that the tumor grows and produces seeding in the eye complicated by retinal detachment, necrosis and invasion of the optic nerve, orbit and the central nervous system [15]. The primary role of a radiologist in the management of a child with a retinoblastoma is to determine the spread of the tumor (optic nerve infiltration, skeletal breakthrough and to determine if there is metastasis to the liver, lymph nodes and to the meninges [2]. In most instances depending on the availability of resources most radiologists avoid performing computed tomography (CT) scan in patients with retinoblastoma due to long-term increased risk for malignancy and that makes the magnetic resonance imaging (MRI) scan the better tool for diagnosing and for the staging of a retinoblastoma [12] . Some of the MRI findings of a retinoblastoma involve [12] : T1 weighted image: Higher signal intensity compared to the normal ocular fluid T2 weighted image: Hypo-intense lesion compared to the ocular fluid Diffusion weighted image (DWI): Reduced diffusion T1 weighted image with contrast: Enhancing tumor presumable secondary to high cellularity of the tumor. | ||||||
|
Conclusion
| ||||||
|
The diagnosis of a retinoblastoma involves a multi-disciplinary approach and imaging modalities include fundoscopy complimented by ultrasound, computed tomography scan and magnetic resonance imaging scan. Retinoblastoma is commonly diagnosed before the age of five and it is extremely rare after the age of six. Education of the community to sick early medical help may reduce the mortality associated with a retinoblastoma in the developing countries. | ||||||
|
Acknowledgements
| ||||||
|
We are thankful to our radiographers Molopa Moyahabo Tink and Makuka Emilly Mogashoa for retrieving and preparing images for this case study. | ||||||
|
References
| ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions
Luvo Gaxa – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Final approval of the version to be published Bafana Elliot Hlatshwayo – Substantial contributions to conception and design, Revising it critically for important intellectual content, Final approval of the version to be published |
|
Guarantor of submission
The corresponding author is the guarantor of submission. |
|
Source of support
None |
|
Conflict of interest
Authors declare no conflict of interest. |
|
Copyright
© 2015 Luvo Gaxa et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information. |
|
|