|
|
Case Report
| ||||||
| Carbamoyl phosphate synthetase 1 deficiency: First report of this rare metabolic disorder in Kingdom of Bahrain with novel mutation | ||||||
| Z. Alsahlawi1, E. Aljishi2, M. Aljufairi3, H. Naser4, J. Häberle5 | ||||||
|
1Pediatric and Metabolic Disease, Medical Genetics Specialist, PO Box 12- Samlmaniya Medical Hospital Hospital, Pediatric Department, Kingdom of Bahrain 2Pediatric and Metabolic Disease Consultant, Pediatric Department, Salmaniya Medical Complex, Manama – Kingdom of Bahrain 3Pediatric Consultant and Neonatologist – Pediatric Department – Salmaniya Medical Complex – Manama – Kingdom of Bahrain 4Pediatric Intern - Pediatric Department – Salmaniya Medical Complex – Manama – Kingdom of Bahrain 5University Children’s Hospital Zurich, Division of Metabolism and Children’s Research Center, 8032 Zurich, Switzerland | ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article] [Similar article in Pumed] [Similar article in Google Scholar] |
| How to cite this article |
| Alsahlawi Z, Aljishi E, Aljufairi M, Naser H, Häberle J. Carbamoyl phosphate synthetase 1 deficiency: First report of this rare metabolic disorder in Kingdom of Bahrain with novel mutation. Case Rep Int 2018;7:100044CRINTZA2018. |
|
ABSTRACT
|
|
Introduction:
Carbamoyl phosphate synthetase 1 (CPS1) is the first enzyme of the urea cycle, which is the pathway responsible for excess nitrogen detoxification and arginine synthesis. Deficiency of CPS1 will cause severe hyperammonemia often already on the first few days after birth. The majority of patients with CPS1 deficiency have a neonatal presentation with encephalopathy and lethargy due to severe hyperammonemia. These patients usually require intensive care treatment with nitrogen scavenging medication and hemodialysis but despite this, the outcome of these cases is guarded. Case Report: Here we report the first case of CPS1 deficiency in a newborn Bahraini girl who was manifesting on the 3rd day of life with encephalopathy, seizures, and hyperammonemia. She was treated with nitrogen scavenging medication and peritoneal dialysis leading to a decrease of the ammonia level. After starting carglumic acid (Carbaglu), which is the allosteric activator of the urea cycle, the metabolic situation was further improved. Unfortunately, the patient had already sustained severe neurological sequelae and expired after one month. Her diagnosis of a defect in CPS1 was suspected based on the biochemical profile and finally confirmed with the finding of a novel mutation in the CPS1 gene. Conclusion: Hyperammonemia is an acute metabolic emergency and often caused by a UCD that should always be part of the differential diagnosis. We report here the first CPS1 deficiency case, confirmed genetically, in Bahrain with novel mutation. | |
|
Keywords:
Carbamoyl phosphate synthetase 1 deficiency, Urea cycle defect, Hyperammonemia
| |
|
INTRODUCTION
| ||||||
|
Urea cycle disorders (UCD) are a group of diseases that result from deficiency or total absence of enzymes activity, cofactors or transporters needed for excretion of waste nitrogen that result from breakdown of protein. The five catalytic enzymes carbamoyl phosphate synthetase 1 (CPS1), ornithine transcarbamylase (OTC), argininosuccinate synthetase (ASS), argininosuccinate lyase (ASL), and arginase (ARG) in the urea cycle or the cofactor producer N-acetylglutamate synthase (NAGS) are related to these disorders [1]. They are all inherited as autosomal recessive traits except for OTC deficiency which is X-linked. The most common presentation of these defects is in newborns who typically present with somnolence, poor feeding, hyperventilation, and seizures, followed by lethargy and coma, which results from the accumulation of ammonia and other precursor metabolites during the first few days of life. CPS1 deficiency is one of the most severe of the UCDs. Individuals with complete CPS1 deficiency rapidly develop hyperammonemia in the newborn period. Children who are successfully rescued from crisis are chronically at risk for repeated bouts of hyperammonemia [2][3]. For that, treatment of these diseases is focused on measures to reduce hyperammonemia as ammonia is a neurotoxic agent that leads to potentially life-threatening brain edema [4][5]. A range of therapeutic options is available to target either ammonia generation or absorption including reduction of dietary protein, ammonia removal through dialysis (either hemodialysis or, less efficient, peritoneal dialysis), and pharmacological drugs as sodium phenylacetate, sodium benzoate, L-arginine and carglumic acid [6]. We report here the clinical course and diagnosis of the first CPS1 patient identified in the kingdom of Bahrain in order to illustrate the panethnic character of this disease and to increase awareness for rare genetic diseases. | ||||||
|
CASE REPORT
| ||||||
|
Clinical course | ||||||
|
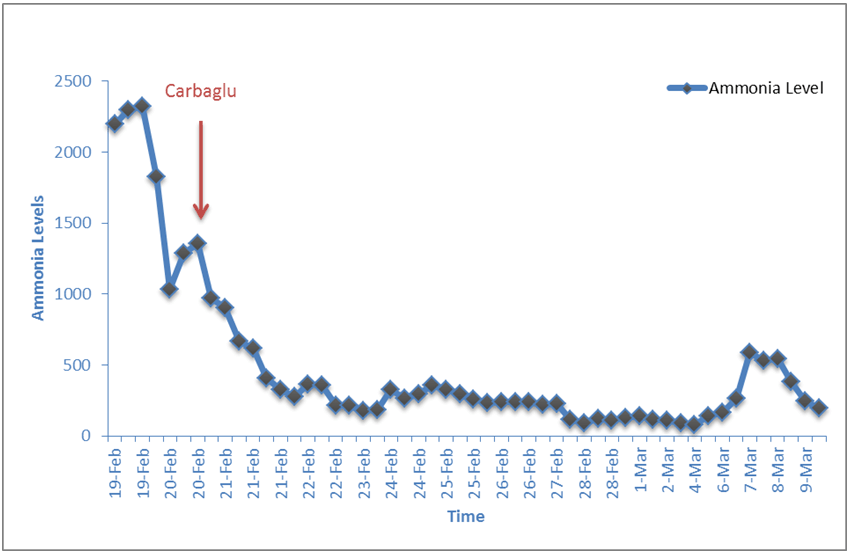
The patient was born at 31 weeks of gestational age due to premature rupture of the membranes for 12 days through normal vaginal delivery with 1.55 kg and an Apgar score of 9/10. The mother was known to have decreased glucose-6-phosphate dehydrogenase (G6PDH) activity and hypothyroidism. The patient was offspring of a consanguineous marriage (parents are second degree relatives). Due to prematurity and to rule out sepsis, the patient was transferred to the neonatal intensive care unit directly after birth and was started on an anti-meningeal dose of ampicillin and gentamicin. On the second day, she was hypoactive, observed to have abnormal body movements consistent with a seizure and was started on phenobarbitone. The following day, she was started on total parenteral nutrition, still hypoactive, developed mottled skin, fixed pupils and arterial blood gas measurement showed respiratory alkalosis. Plasma ammonia was determined and found highly elevated at 2295 µmol/L (normal < 100). Based on this extensive hyperammonemia together with respirator alkalosis, the patient was suspected to have a UCD and was started on intravenous 600 mg/kg L-arginine, intravenous 250 mg/kg sodium benzoate, oral 200 mg carglumic acid (Carbaglu) three times a day, and she received 1.5 times the patient’s maintenance in intravenous fluids, along with peritoneal dialysis, which is the first line extracorporeal detoxification in our institution and the only option given the low birth weight of the patient. The patient also required medication for low blood pressure, and was started on dobutamine, dopamine and hydrocortisone, which were all stopped after her condition started to improve. Peritoneal dialysis was done for two days, and ammonia levels started decreasing over a period of five days finally reaching 176 µmol/L (Figure 1). Oral sodium phenylbutyrate 250 mg/kg per day was also introduced at that point. On the next day, ammonia was normal (65 µmol/L), L-carnitine was introduced to cover the possibility of a secondary carnitine deficiency, and total parenteral nutrition (TPN) with aminoplasmin 0.5 g/kg/day was started.
Tandem mass spectrometry (TMS) showed a low citrulline level (1.83 µmol/L, normal: 3–70) and a normal acylcarnitine profile. Thus, there was no indication for an organic acidemia but the results were still suggestive of a UCD. Since orotic acid in urine was not increased, the defect was thought to be either CPS1 or NAGS deficiency and L-carnitine was stopped. When ammonia levels went up again to 230 µmol/L, treatment was adjusted (L-arginine was reduced to 300 mg/kg per day and Aminoplasmin was increased to 1 g/kg per day) aiming to withdraw some of the scavengers intravenous medication and to advance her nutritional intake. At 15 days of age, the patient was metabolically stable with normal ammonia levels, and was thus started on a maintenance therapy including a low-protein diet, supplementation of essential amino acids, and oral treatment with Carbaglu 200 mg divided in three doses per day, arginine 250 mg/kg/day, and sodium benzoate 250 mg/kg/day. Cranial ultrasound showed no signs of brain edema, interventricular hemorrhage or any other abnormalities. The patient was experiencing recurrent attacks of seizures, which were followed with the neurological team to adjust her antiepileptic medication, and she was observed with cerebral function monitoring (CFM) showing continuous abnormal sharp waves, which was interpreted as poor prognostic feature. At 21 days of age, molecular genetic investigation was sent to confirm a proximal UCD. Concomitantly, the patient’s condition was discussed with parents and a no resuscitation status was agreed upon. A few days later, patient suddenly desaturated, and expired at 33 days of age. | ||||||
| Genetic analysis | ||||||
|
Mutation analysis was done using 1 ml heparin blood from the patient following a published protocol [7]. In brief, heparin full blood was cultured for four days in the presence of phytohemagglutinin and cycloheximide followed by RNA-isolation and cDNA-synthesis. Amplification of the CPS1 transcript was done by PCR followed by direct sequencing of the CPS1 transcript in six overlapping fragments. Hereby, the novel homozygous mutation c.1812_1813del (p.Glu604Aspfs*31) was found and confirmed in DNA. Both parents were found to be carriers of this mutation. | ||||||
| ||||||
|
DISCUSSION | ||||||
|
Carbamoyl phosphate synthetase 1 deficiency is a rare disorder of the urea cycle that results from a deficiency in the first enzyme of the pathway that detoxifies ammonia. The disorder can present in two forms that are part of a disease continuum, neonatal lethal form and less severe late onset form [8][9]. The lethal form was first reported in a patient with congenital hyperammonemia and decreased levels of CPS [10]. Our case likewise presented a typical lethal neonatal form as the patient presented at day-2 of life with severe hyperammonemia, encephalopathy and seizures. The management of the patient was done following standard recommendations including detoxification of the acute severe hyperammonemia through dialysis to rapidly reduce the plasma ammonia concentration; intravenous administration of L-arginine hydrochloride and nitrogen scavenger drugs to allow alternative pathway excretion of excess nitrogen; restriction of protein for 12–24 hours to reduce the amount of nitrogen in the diet; calories given as carbohydrates and fat; and physiologic stabilization with intravenous fluids and cardiac pressors while avoiding overhydration. In our patient, who received all of the aforementioned, serial ammonia testing showed partial improvement after L-arginine and sodium benzoate but a more pronounced reduction after introducing carglumic acid. Diagnosis of a urea cycle disorders in general and CPS1 deficiency in particular is established based on the clinical presentation, family history, and analysis of plasma amino acids and urinary orotic acid. Confirmation is best done using mutation analysis. In our case, plasma amino acids showed a low citrulline, no increase in urinary orotic acid, and high ammonia with encephalopathy. Thus, CPS1 deficiency was suspected and finally confirmed when the novel mutation c.1812_1813del (p.Glu604Aspfs*31) in CPS1 was found. This mutation is a deletion of a single nucleotide leading to frameshift and early stop of translation. Thus, is has to be assumed that patients with this mutation are virtually lacking any active CPS1 protein. In agreement with this, the neonatal presentation of CPS1 deficiency in our patient was very severe despite of the intensive care management. Although this treatment was efficacious since ammonia levels initially normalized, the patient sustained severe neurological symptoms with continuous seizures, and finally expired at around one month of life. This can be best explained by the high ammonia levels at the very beginning that probably have led to irreversible brain damage. | ||||||
|
CONCLUSION
| ||||||
|
Hyperammonemia is an acute metabolic emergency and often caused by a urea cycle disorders that should always be part of the differential diagnosis. We report here the first carbamoyl phosphate synthetase 1 (CPS1) deficiency case, confirmed genetically, in Bahrain illustrating that CPS1 deficiency is panethnic and potentially life-threatening. Nevertheless, knowledge about the exact molecular background allowed family counseling and hopefully helped the family to cope with the very sad clinical course. | ||||||
|
REFERENCES
| ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions
Z. Alsahlawi – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published E. Aljishi – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published M. Aljufairi – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published H. Naser – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published J. Häberle – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published |
|
Guarantor of Submission
The corresponding author is the guarantor of submission. |
|
Source of Support
None |
|
Conflict of Interest
Authors declare no conflict of interest. |
|
Copyright
© 2018 Z. Alsahlawi et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information. |
|
ABOUT THE AUTHORS
| |||||||||||||||
| |||||||||||||||
|
|